

Аннотация
Proteomic methods and approaches to the detection of tissue-specific and tissue-generating proteins and peptides – which form corrective properties – in studied meat samples and specially developed meat products were successfully tried out in 2016–2017. The methods allow one to confirm protein and peptide authenticity and also detect bio-markers of proteolytic changes in meat after slaughter. The following proteomic techniques were used in the present research: two-dimensional O’Farrell electrophoresis with isoelectrofocusing in ampholin and immobilin pH gradients, the detection of proteins on two-dimensional electrophoregrams by staining with Coomassie R-250 and silver nitrate, and mass spectrometric identification of proteins by means of MALDI-TOF and MS/MS methods. Contractile actomyosin complex proteins, such as myosin light chains and tropomyosins, were the most informative among proteins of species specificity. It is also necessary to mention that earlier experiments allowed us to choose enzymes which play a part in carbohydrate metabolism (glyceraldehyde 3-phosphate dehydrogenase and β-enolase) as markers. In addition to the listed proteins, myoglobins, actins, and several other proteins in horse meat have showed high species specificity and have been detected well. A system of species specificity (authenticity) of meat raw materials was suggested. The system allows the presence of pork, beef, horse, and camel meat to be detected in both raw and heat-treated products if the content is 5% and more. The data has been obtained by means of bioinformatics, а highly useful tool for formulating an algorithm to identify the protein markers for the Atlas “Proteomic profiles of farm animals meat proteins”. “Proteomic profiles of farm animals muscle proteins”.Ключевые слова
Beef, pork, horse meat, camel meat, proteomics, muscle proteins, peptide fingerprint, 2D-electrophoresis, MALDI-TOF mass spectrometry, authenticityВВЕДЕНИЕ
During last 10 years, scientists throughout the world have been researching protein and peptide substances in raw and processed meat products, which are formed as a result of technological treatment and bring about the quality, functionality, and safety of final food products. Peptides containing 2–30 amino acids have been defined. The peptides have hypotensive, opioid, antioxidant, antimicrobial, and other biological effects on a number of the most common mechanisms underlying various pathological processes. The investigation of meat proteins as potential sources of biopeptides includes the study of both proteome and metabolites formed due to the fermentation process as well hydrolysis of raw materials by gastrointestinal tract natural enzymes by means of proteomics and bioinformatics [1, 3]. The presence of hypothetical functional sequences and the formation of active sequences due to the action of own proteolytic enzymes, proteases and peptidases of bacterial origin were studied.
Numerous researches in the world practice are concerned with the study of the biological role of peptides in vivo, their absorption and resistance to gastrointestinal tract enzymes. Mechanisms of protein and peptide formation which bring about bio corrective and qualitative characteristics, as well as biosynthesis, folding, and catabolism processes are of great interest.
Bioinformatics as the tool of studying proteome in relation the hypothetical presence of various bioactive peptides and protein makers in it is becoming more and more popular. According to available data, muscle proteins of food-producing animals contain amino acid sequences with various biological properties such as beef collagen α1, myosin light chain (LC), and connectin [2]. Beef collagen α1 has hypotensive, opioid, anti-amnesic, and antithrombotic properties and stimulates ubiquitin-regulated proteolysis which inhibits dipeptidyl peptidase IV and adjusts gastric mucosa activity; мyosin LC is rich with antimicrobial sequences; connectin contains peptides of antithrombotic, anti-amnesic, opioid, neuroprotective, immunomodulatory, antioxidant, and hypotensive activity, dipeptidyl peptidase IV inhibitors, and regulators of gastric mucosa activity. Beef, chicken, and pork actins contain inhibiting dipeptidyl peptidase IV sequences. Collagen and elastin include sequences with corrective properties due to high glycine and proline content. It should be noted that over 220 functional peptides have been identified in the above-mentioned proteins [4–6].
It should be noted that the replacement of the basic component in the product, even partially, can have an adverse effect on the product functionality; not only does nutritional value tend to increase but also biological value. In such case, proteomics is of help.
Proteomics is aimed at the identification of all the proteins, their biological activity, post-translational modifications, cell processes, and changes in “proteome” as a reaction to changed biological conditions. The typical sequence of operations in proteomics includes extraction and separation of proteins/peptides, their identification, and data analysis. The most common method used to determine proteins or peptides in proteomics is mass spectrometry. The strategy is fairly applicable in many spheres; however, it is limited by the great biochemical heterogeneity of proteins and impossibility of accurate determination of less common proteins [7].
During recent decades, proteomic methods with high output are developing and improving rapidly, which has considerably changed experimental approaches for food science. The interest in using genomics, proteomics, and metabolomics in the science of meat to obtain useful information about meat characteristics is growing. Proteomics takes an important part in life sciences, including agriculture, food and animal sciences to make safe food products of high quality and improve ecological rationality of livestock farming [3]. Such important parameters as meat composition, sensory quality, and nutritional value are responsible for meat quality and its acceptability for customers. Meat quality is closely related to animal biological characteristics. Meat quality parameters such as delicacy, water-binding capacity, fractional content, autolytic changes, and others are complicated multi-component indicators; hence they would be characterized in detail on the basis of experimental approaches and techniques aimed at parallel studies of genes and proteins simultaneously.
Proteomics is a promising approach to study mechanisms underlying the meat quality and the effect of meat on human health.
Proteomics is aimed to identify molecular markers, usually named bio-markers, which allow earlier and more accurate diagnostics of diseases in medicine, for instance. Currently, bio-marker search is of importance since bio-markers may be used to improve the great number of characteristics in meat production and processing.
The aim of the research was the integration of existing knowledge and its practical application to identify the protein/peptide markers by means of proteomic and bioinformatic methods on the basis of their proteomic profiles which confirm or refute claimed properties of the meat product.
ОБЪЕКТЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Experimental studies were conducted at “Scientific and Methodological Works, Biological and Analytical Researches Laboratory” of “Gorbatov Research Center for Food Systems” in collaboration with the “Protein Research Laboratory” of the Federal Research Center of Biotechnology of the Russian Academy of Sciences.
Subjects of the study in identifying specific peptide markers were myofibrillar and sarcoplasmic proteins, as they are presented in meat in large amounts; thus, the method can be expected to be highly sensitive. Moreover, these proteins are completely soluble in buffer. For the experiment, average samples of longissimus dorsi from chilled pork and pork after 5 days of autolysis, as well as cooled beef and horse meat were used. Among the specific peptide markers for each animal species, the most sensitive peptides were selected from a protein database to develop the high performance liquid chromatography method (HPLC) in combination with tandem mass spectrometry.
For protein extraction, 300 g of each kind of meat – beef, pork, horse, and camel – finely chopped were mixed with 4 ml of the extraction buffer (0.3 M of KCl, 0.15 M of KH2PO4, and 0.15 M of KH2PO4 with pH 6.5) and kept for two hours at room temperature with constant stirring by shaking. Then the samples were subjected to centrifugation for 60 minutes at 12,000 g and 40°C. 100 µm of the supernatant was evaporated in a stream of nitrogen at 39°C and placed into 100 µm of 6 molar urea. After reduction with dithiothreitol (DTT) and alkylation with iodide acetamide (IA), cleavage process was running on exposure to trypsin for overnight at 37°C with slow stirring by shaking. The samples were then diluted with deionized water in a ratio of 1 : 2 and desalted by using Strata-X (30 mg). For this, 1 ml of a mixture consisting of 5% methanol and 1% formic acid was washed with water. For the elution of the peptide mixture, 1 ml of methyl cyanide/water (90 : 10; 0.1% formic acid) was used. The eluate was placed into Eppendorf tubes with 5 µm of dimethylsulphoxide (DMSO). Two-dimensional O’Farrell electrophoresis with isoelectrofocusing in ampholin (IEF-PAGE) and immobilin (IPG-PAGE) pH gradients and detection of proteins on two-dimensional electrophoregrams by staining with Coomassie R-250 and silver nitrate were used as main proteomic techniques [1, 13].
Protein fractions selected for the identification were cut out of gel plates obtained by means of twodimensional electrophoresis. Gel sections were cut out, the protein was hydrolyzed with trypsin, and then tryptic peptides were extracted using time-of-flight mass spectroscopy on the matrix (MALDI-TOF) in accordance with earlier published techniques [8, 9] with some modification [10]. The degree of extraction was determined by the identification of model proteins with known weight (PageRuler Protein Ladder, a mixture of 14 recombinant proteins with molecular weight from 10 to 200 kDa). The protein yield in the experiment was from 88 to 94%.
The study sample (0.5 µm) was mixed with the same volume of 20% methyl cyanide containing 0.1% of triflouracetic acid and 20 mg/ml of 2,5-dihydroxybenzoic acid (“Sigma”, USA) and subjected to air drying.
The mass spectrometric identification of the peptides was carried out after the separation of the mixture by HPLC method on the RP18 column by using MALDITOF MS and MS/MS spectrographic methods; for this, a MALDI-TOF time-of-flight mass spectrometer Ultraflex (“Bruker”, Germany) with UV laser (336 nm) in the positive ion mode within the weight range from 500 to 8,000 Da was used. The peptides were calibrated with known peaks of trypsin autolysis.
At least three MRM transitions (transitions in mode of multiple reaction monitoring) were selected for more accurate identification. Mass spectrometric parameters optimization was carried out from the study of peptide extracts or synthetic peptides. Various matrices were investigated to verify the specificity of the peptide markers found in the database and to eliminate false results; this is of importance because of the proteomic data Imperfection.
Proteins were identified by using peptide maps from the protein sequences database of NCBI (the National Center of Biotechnological Information) and the Mascot Software (http://www.matrixscience.com). Initial parameters of the search included one missed cleavage site in tryptic peptides, carbamidomethylation of cysteine, partial oxidation of methionine, and a mass-to-charge ratio discrepancy (m/z) about 25 ppm.
For benchmarking the proteomic profiles, modules of UniProtKB/the Swiss Institute of Bioinformatics and the database “Skeletal muscle proteomics” (http://mp.inbi.ras.ru) were used [11].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
The fractionation of the protein extractions from beef, pork, and horse longissimus samples by means of 2D-electrophoresis with isoelectrofocusing in ampholin pH gradient (IEF-PAGE) ensured a large number of protein fractions when staining with Coomassie R-250 [12]. The number of these fractions was determined automatically on digital images of twodimensional electrophoregrams (2-DE) using ImageMaster 2D Platinum, version 7 (“GE Healthcare”, Switzerland). It should be noted that the total distribution of protein fractions detected was similar to that of muscle proteins in meat of earlier studied animals. Fig. 1 demonstrates the similarity when comparing major fractions for tropomyosins and myosin light chains (MLC).

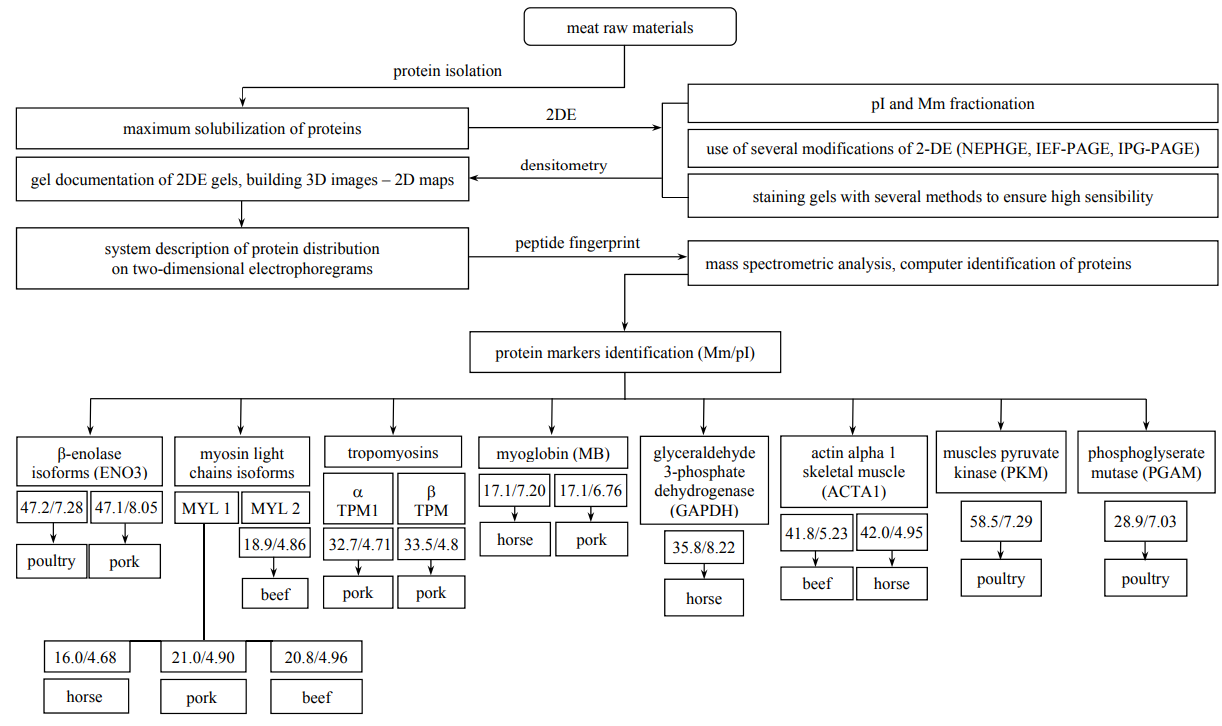
Proteins of contractile actomyosin complex, such as myosin light chains and tropomyosins, were selected as the most informative among species-specific proteins. Also, according to earlier experiments, enzymes which take a part in carbohydrate metabolism (glyceraldehydes 3-phosphate dehydrogenase, β-enolase) have been selected as markers. In addition to the listed proteins, myoglobins, actins, and several other proteins in horse meat have had high species specificity and have been detected well.
On the basis of data obtained in both present and previous experiments [13, 14], the system of species specificity (authenticity) of meat raw materials was suggested (described below). The system allows the presence of pork, beef, horse, and camel meat to be detected in both raw and heat-treated products if the content is 5% and more.
More detailed ways of identification are presented below. Thus, Fig. 2 shows 2-DE of average samples of pork longissimus and coupling.

Multi-year proteomic researches of different species of meat raw materials (pork, beef, horse, and camel) as well as cooked meat products (sausages) demonstrate fractions which were identified as known tissue-specific proteins to present as major proteins: α- and β-tropomyosins (1), myosin light chains 1 MLC and MLC 2 (2 and 3), and myoglobin (4).
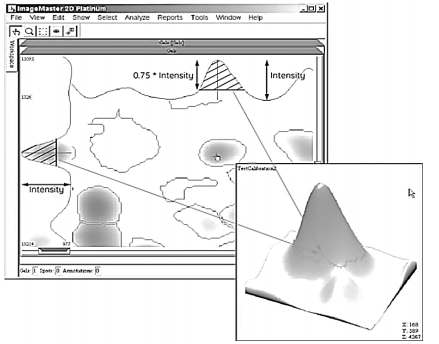
The number of muscle proteins was determined automatically on digital images of two-dimensional electrophoregrams (2-DE) using ImageMaster 2D Platinum, version 7 (“GE Healthcare”, Switzerland). The software finds stained spots and draws a contour over the stained area. The intermediate result of this analysis is represented in Figure 3.

Further, mathematical algorithms were used to determine a difference between light and dark pixels. Since the spots have different degree of staining and can blend into the background, the program sets three special parameters to identify protein fractions (spots). One of them (“Smooth”) enables the most marked spots identification and sharpness of the spot edge detection. The second parameter (“Saliency”) is used to identify weakly stained spots from the background and noise. “Min Area”, the third parameter, sets the minimum area of the spot; thus, all the spots with less area are not analyzed.
The next stage of the image processing includes the collection of information about spots and 3D models construction. The analysis involves such parameters as intensity, area, and volume of spots.
The intensity shows a degree of staining compared to the background; for this, the values of the most strongly stained pixels of a spot and the lightest area of the nearest background surrounding the spot are taken.
The spot area (in mm2 ) is calculated from a mean value of the staining intensity. The spot edge determination in automatic mode includes some difficulties, therefore slightly larger area is assigned to the spots, and the index value is taken as 75% from the spot intensity. The area is expressed in mm2.
The spot volume reflects the integrated optical density of the study fraction and is computed from the spot area magnitude which in turn is calculated strictly along the spot outline.
The final models represent sets of peaks; the higher intensity, the higher the peak and the larger the protein concentration in the fraction. The peak height is taken as 75% from the spot intensity [15].
Basic steps in the 3D model constructing are demonstrated in Fig. 4. The final result is shown in Fig. 5.

Thus, computer densitometry techniques are successfully applicable to use the species-specific protein markers in estimating a quantity and a type of protein in meat raw materials and meat products; correction factors can be used to take into consideration different protein content in raw materials.
On the basis of the data about the real proportion of meat raw materials in meat products produced according to standard formulations in study pork/beef samples (Fig. 6) and the confirmed species-specific bio-markers (through the example of myoglobin), the proper correction factor (KC) was calculated. Its value turned out to equal 0.67 OD/C units to determine the major protein in pork (here, OD is the value of optical density of pork myoglobin fraction measured by means of computer densitometry, and C is the protein content in units). For beef myoglobin, KC was determined and had the value of 1.5 OD/C units.

After correction of the results by using KC, a ratio of pork/beef was 56.3/43.7 for sample No. 1, 57.4/42.6 for sample No. 2, and 57.6/42.4 for sample No. 3 (in %).
Summarily, a ratio for the three samples was 57.1/42.9 that almost coincided with the results of the three other species-specific markers (Fig. 5).
The technique developed for the detection of species specificity of meat raw materials is highly useful to determine meat product composition and the presence/absence of components. On the other hand, the technique has some limitations in determining quantitative values by reason of different physical and chemical properties of peptides, which directly affect their detection.
Myoglobins of horse/beef/pork, for example, show discrete spots differing in Mm and pI that allows their presence on obtained elecrophoregrams to be determined visually but it is not easy to differentiate horse myoglobins from that of beef due to their high homology and similar electrophoretic characteristics. Therefore, we decided to use isoforms of β-enolase and muscle actin alpha instead of myosins as bio-markers in meat raw materials because of their higher species specificity, which means that they are detectable and identified well.
The data obtained in the present and previous experiments [16] were organized using bioinformatics, which has been highly useful for formulating an algorithm to identify the protein markers (Fig. 7) and expand the Atlas “Proteomic profiles of farm animal meat proteins”. Fig. 7 demonstrates all the detected species specificity protein markers in meat raw materials.
This investigation is also useful for meat-packing factories which use a variety of raw materials in single shift. The development of criteria for classifying product components as technologically inseparable impurities is a challenge with large-scale monitoring. First, undeclared components being contained in production must be shown on labels.
Statements on the label “may contain insignificant amount…”, “contains trace amounts…”, and others do not give accurate criteria whether the declared component is technologically inseparable impurity or intentionally introduced into the product. This fact can lead to deliberate substitution of a small part of ingredients in the formulation (for example, the substitution of meat raw materials by vegetable protein, in particular, soya) without declaring the ingredient in the composition, but with the statement “may contain insignificant amount…”, “contains trace amounts…”, and others. Since the value of “insignificant amount” is not regulated by standards, dishonest manufacturers do not bear administrative liability.
The practical application of the developed algorithm in the future will involve work on comparative interpretation of the detected proteins which will be aim at finding deviations from technological formulas. Some aspects and primary results will be given in this paper.
To understand the autolysis process, it is necessary to know an effect of changes in muscle fiber and the cells of muscle fiber on muscle proteins. At the same time, it should be noted that these changes have been little investigated nowadays. Obviously, it is autolysis that leads to the breakdown of proteins – in particular, specific myofibrillar proteins and proteins of cytoskeleton, titin, and nebulin – and the formation of peptides. The protein breakdown begins in 6 hours after slaughter [17].
Nevertheless, it is not completely clear what factors cause the breakdown – own meat enzymes, or fragmentation as a result of amino acid modification chemically influenced [18], or direct hydrolysis caused by weak acid and acidic medium in muscles tissues.
The results of the identification (Table 1) showed that the most striking feature of autolysis is the formation of fast skeletal muscle troponin T fragments. The presence of ordinary troponin fraction caused the appearance and growth of three additional fragments with different Mm and pI. Increased amount of such protein fragments as piruvate kinase, α-enolase, creatine kinase M-type, glyceraldehydes-3=-phosphate dehydrogenase, troponin I, adenylate kinase, alpha-cristallin, myosin light chains, and cofilin were observed.
Knowledge of protein changes during processing and the variability of the results will be of value in the improvement of treatment process. Numerous studies showed that variations in the rate of post-mortem glycolysis allow the meat of different delicacy to be obtained [19].
However, future studies are necessary for clearer understanding of complicated mechanisms of postmortem changes.
ВЫВОДЫ
In conclusion, the obtained results of proteomic studies of farm animals and poultry muscles is today a valuable contribution in developing highly sensitive techniques for the quality control of meat products based on analyses of muscle proteins species-specific isoforms.
In the beginning of the 21st century, proteomic and bioinformatic techniques had taken a significant part in protein biochemistry [6]. Accumulated results of numerous researches represent information arrays on the basis of which various common and specialized databases are formed and placed in the internet. Among them, the informational resource in the database UniProt called “Completeproteom of Homosapiens” should be particularly noted. It included more than 70,000 papers by middle of 2012; however, only 25,000 (35.2%) of them represented the results of direct studies of the corresponding protein. Thus, it is evident that the majority of the papers (64.8%) require additional experimental data. Proteomic techniques and the development of informatics resources can play a significant role here, which indicates high topicality of such researches.
Along with studying different aspects of protein polymorphism and other fundamental problems of muscle protein biochemistry, proteomic technologies have the tendency to be widely used in solving biomedical problems directed at a number of applied tasks – from detecting potential diagnostic protein polymers, targets for pharmacological interventions, to developing methods of quality control of various food products produced from animal muscle tissue [19].
The final stage in studying proteomic protein profiles for many scientists is the obtaining of twodimensional electrophoregrams, as they do not have any considerations how to use modern instrumental and bioinformatic resources to confirm/refute their hypotheses or even just to identify.
The data obtained in the present and previous experiments have been organized, proteomic and bioinformatic methods of studying protein markers have been applied practically, and the identification algorithm of species-specific proteins in slaughter farm animals and poultry meat – which makes it possible to confirm the authenticity of raw materials – has been developed.
In future research, we are planning to carry out work on comparative interpretation of the identified proteins in order to apply in practice the developed foundations and the identification algorithm aimed at detecting deviations from technological formulas and predicting functional and technological properties of meat products. At present, these tasks are of importance for autolytic processes studying of raw materials under the conditions of changing animal genotype and forage base.
The is financially supported by the Russian Science Foundation (project No. 16-16-10073).
СПИСОК ЛИТЕРАТУРЫ
- Shishkin S.S, Kovalev L.I, Kovaleva М.А., et al. The application of proteomic technologies for the analysis of muscle proteins of farm animals used in the meat industry (Review).Applied Biochemistry and Microbiology, 2014, vol. 50, no. 5, pp. 453–465. (In Russian).
- Vostrikova N.L. and Chernukha I.M. Bioinformatics – instrument interpretation proteomic profiles of meat protein. Theory and practice of meat processing, 2017, vol. 2, no. 1, pp. 4–17. (In Russian). DOI: 10.21323/2414-438X-2017-2-1-4-17.
- 3. Chernukha I.M., Fedulova L.V, Kotenkova E.A., Shishkin S.S., and Kovalyov L.I. The Influence of Autolysis on the protein-peptide profile of Bos taurus and Sus scrofa Heart and aorta Tissues. Theory and practice of meat processing, 2016, vol. 1, no. 2, pp. 4–9. (In Russian). DOI:10.21323/2414-438X-2016-1-2-4-9.
- Picard B., Lebret B., Cassar-Malek I., et al. Recent advances in omic technologies for meat quality amangement. Meat Science, 2015, vol. 109, pp. 18–26. DOI: 10.1016/j.meatsci.2015.05.003.
- Zhang R., Große-Brinkhaus C., Heidt H., et al. Polymorphisms and expression analysis of SOX-6 in relation to porcine growth, carcass, and meat quality traits. Meat Science, 2015, vol. 109, pp. 18–26. DOI: 10.1016/j.meatsci.2015.04.007.
- Anderson N.L., Polanski M., Pieper R., et al. The Human Plasma Proteome: A Nonredundant List Developed by Combination of Four Separate Sources. Molecular & Cellular Proteomics, 2004, vol. 3, no. 4, pp. 311–326. DOI: 10.1074/mcp.M300127-MCP200.
- Shishkin S.S., Kovaleva M.A., Eryomina L.S., Lisitskaya K.V., and Kovalev L.I. Proteomic Approaches for the Study of Transgelins as Tumor-associated Proteins and Potential Biomarkers. Current Proteomics, 2013, vol. 10, no. 2, pp.165–178. DOI: 10.2174/1570164611310020008.
- Granvogl B., Plöscher M., and Eichacker L.A. Sample preparation by in-gel digestion for mass spectrometry-based proteomics. Analytics and Bioanalytics Chemistry, 2007, vol. 389, no. 4, p. 991–1002. DOI: 10.1007/s00216-007-1451-4.
- Medzihradszky K.F. In-solution digestion of proteins for mass spectrometry. Methods in Enzymology, 2005, vol. 405, pp. 50–65. DOI: 10.1016/S0076-6879(05)05003-2.
- Zvereva E.A., Kovalev L.I., Ivanov A.V., et al. Enzyme immunoassay and proteomic characterization of troponin I as a marker of mammalian muscle compounds in raw meat and some meat products. Meat Science, 2015, vol. 105, pp. 46–52. DOI: 10.1016/j.meatsci.2015.03.001.
- SIB Swiss Institute of Bioinformatics. Available at: http://web.expasy.org/docs/swiss-prot_guideline.html/ UniProtKB/Swiss-Prot/SIB Swiss Institute of Bioinformatics. (accessed 10 May 2017).
- Anderson N.L. and Anderson N.G. Proteome and proteomics: New technologies, new concepts, and new words. Electrophoresis, 1998, vol. 19, no. 11, pp. 1853–1861. DOI: 10.1002/elps.1150191103.
- Kovalev L.I., Shishkin S.S., Kovaleva М.А., Vostrikova N.L., and Chernukha I.М. Proteomic research proteins in a sample of pork meat products. All about the meat, 2013, no. 3, pp. 32–34. (In Russian).
- Manyukhin Ya.S., Chernukha I.М., Kovalev L.I., et al. The study of horsemeat proteins by use proteomic technologies. All about the meat, 2014, no. 3, pp. 20–24. (In Russian).
- Ivanov А.V. Sravnitel’noe proteomnoe issledovanie belkov cheloveka, uchastvuyush’ikh v obespechenii dvigatel’nykh funktsiy [Comparative proteomic study of human proteins taking part in locomotor functions]. Abstract of Diss. Cand. Sci. (Eng.). Moscow, 2012. 25 p.
- Manyukhin Ya.S., Chernukha I.М., Vostrikova N.L., et al. The study of muscle proteins of camel using proteomic technologies. All about the meat, 2016, no. 6, pp. 24–28. (In Russian).
- Melody J.L., Lonergan S.M., Rowe L.J., et al. Early post mortem biochemical factors influence tenderness and water-holding capacity of three porcine muscles. Journal of Animal Science, 2004, vol. 82, no. 4, pp. 1195–1205.
- Stadtman E.R. Metal ion-catalyzed oxidation of proteins: Biochemical mechanism and biological consequences. Free Radical Biology and Medicine, 1990, vol. 9, no. 4, pp. 315–325. DOI: 10.1016/0891-5849(90)90006-5.
- Lametsch R. Proteomics in Muscle-to-Meat. Proceedings of the American Meat Science Association 64th Reciprocal Meat Conference. Kansas, 2012, pp. 19–23.
- Jira W. Aktuelles aus der internationalen Fleischforschung massenspekrtrometrischer Nachweis von Tierarten und Klebefleisch. Fleischwirtschaft, 2014, vol. 94, no. 5, pp. 98–101.
- Pares D., Saguer E., Pap N., Toldra M., and Carretero C. Low-salt porcine serum concentrate as functional ingredient in frankfurters. Meat Science, 2012, vol. 92, no. 2, pp. 151–156. DOI: 10.1016/j.meatsci.2012.04.029.
