

Аннотация
Introduction. Barley can be infected with a broad variety of fungi, which can cause considerable loss of crop yield and reduce the quality of grain. Modern vision on the geographical and ecological distribution and biodiversity of micromycetes has been established by traditional, cultivation-based methods. However, more recently, molecular methods have shifted microbiological research to a new level, making it possible to investigate hidden taxonomical biodiversity.Study objects and methods. For this study, we determined the fungal biome on the surface and inside of barley grains using the traditional mycological method and the contemporary molecular method, which employed DNA metabarcoding based on NGS (nextgeneration sequencing) of the ITS2 region. We analyzed five cultivars that were collected in two subsequent crop seasons (2014, 2015).
Results and discussion. DNA metabarcoding revealed 43 operational taxonomic units, while 17 taxa of genus or species level were recovered by the traditional method. DNA metabarcoding revealed several minor species and one predominant, presumably plantpathogenic Phaeosphaeria sp., which were not detected in the agar plate-based assay. Traditionally, Fusarium fungi were identified by mycological assay. However, the resolution of DNA metabarcoding was sufficient to determine main Fusarium groups divided by ability to produce toxic secondary metabolites. The combined list of Ascomycetes consisted of 15 genera, including 14 fungi identified to species level. The list of Basidiomycota derived from DNA metabarcoding data alone included 8 genera.
Conclusion. It was found that crop season predetermines the fungal community structure; mycobiota on the surface and inside of grain was significantly different.
Ключевые слова
Barley, seed-borne fungi, infection, next-generation sequencing, rDNA, Alternaria, FusariumВВЕДЕНИЕ
Barley (Hordeum vulgare L.) is one of the major cereal crops. It occupies fourth place among cereals in the world and second place in Russia by production quantity and cultivation area [1]. The importance of barley has been accepted since ancient times and used in the food, feed and brewing industries due to its versatility, excellent adaptation capabilities and superior properties [2].
The increased interest in barley as a source of food and fodder has resulted in a huge number of studies of associated microorganisms. It is known that barley can be infected with a broad variety of plant-pathogenic and toxigenic fungi, many of which may persist in grains. The Bipolaris, Pyrenophora, Phaeosphaeria, Alternaria, and Fusarium genera are considered to be prevailing fungi in barley grain worldwide [3, 4]. Species of the last two genera are well known as mycotoxin producers, with Fusarium spp. being the most dangerous food and feed contaminants.
Cultivation-based methods have traditionally established modern vision on geographical and ecological distribution and biodiversity of micromycetes. These methods cannot provide accurate data on taxon composition because some microorganisms do not have specific characteristics to be identified, and some appear to be noncultivable. Thus, data on the mycobiome of many substrates, including barley grain, is likely to be incomplete.
In recent decades, molecular methods have shifted microbiological research to a new level, making it possible to investigate hidden taxonomical biodiversity. Next-generation sequencing (NGS), implemented on various independent technical platforms, became the most promising method for conducting research projects aimed at revealing fungal or bacterial composition [5–7]. Several studies have focused on a variety of agricultural subjects [8–12]. Advances in this field led to consideration that NGS-based methods are suitable as incipient techniques for seed testing [13].
In Denmark, 454 pyrosequencing of the internal transcribed spacer 1 (ITS1) of the nuclear ribosomal DNA (rDNA) has been chosen to recover the composition of fungal communities associated with wheat grain [14]. NGS revealed a significantly higher level of biodiversity than it was observed in previous culturing studies. Another appropriate 454 pyrosequencing of both ITS regions was done to study the mycobiome of barley grain in western Canada [3]. It demonstrated that geographic location and agronomical practices were the determining factors explaining the observed differences in the fungal communities associated with barley. Such studies may contribute to a better understanding of fungal species compositions in cereals. They may also lead to more accurate food-quality testing and the precise design of crop protection strategies that would reduce the level of fungal contamination of agricultural products.
The objective of this study was to revise the taxonomical variety of fungi contaminated the surface and infected barley grains harvested in the northwestern region of Russia. We hypothesized that grain mycobiome could be significantly differ on the surface and inside the grain, and the difference may depend on the crop year. In our research we used the traditional agar platebased method and the contemporary method based on 454 pyrosequencing of ITS2.
ОБЪЕКТЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Sampling. Grain samples of five spring barley cultivars (Suzdalets, Krinichnyy, Moskovskiy 86, Tatum, and Belgorodskiy 110) were received in 2014 and 2015 from the State Experimental Station (Volosovo, Leningradskaya oblast, Russia, 59°31’N, 29°28’E). Small grain cereals on this station were cultivated with no fungicide treatments. The grain samples intended for fungi isolation and DNA extraction were stored separately at 4°C and –20°C respectively.
Extraction of DNA. Two representative subsamples consisting of 500 grains were picked from each sample. One subsample was placed in a 50 mL polypropylene tube and subjected to superficial sterilization. The grains were consistently washed with 20 mL of 2% sodium hypochlorite solution containing 0.1% of sodium dodecyl sulphate (SDS). They were then washed once with 5% sodium hypochlorite, two times with deionized water (diH2O), and finally rinsed with 98% ethanol. With each step the mixtures were actively stirred up within 2 min, and the flushing solution was then decanted without the grain. The ethanol was removed by burning at regular stirring during 10 s. After this, the grains were homogenized in sterile disposable chambers on a Tube Mill Control (IKA, Germany) grinder. The other subsample was similarly homogenized, but the step of superficial sterilization was skipped.
Further, 240 mg of the ground subsamples were transferred to a 2 mL Eppendorf tube, where DNA was extracted with an AxyPrep Multisource Genomic DNA Miniprep Kit (Axygen, USA), according to the centrifugal protocol for plant tissues and fungal mycelium. The DNA concentrations were measured with a Qubit 2.0 (Thermo Fisher Scientific, USA) using a dsDNA HS Assay Kit. Extracted DNA was used for library preparation and subsequent universal tailed amplicon sequencing, as described for the 454 Sequencing System.
Pyrosequencing and primary data analyses. For amplicon library preparation we chose the taxonomically significant ITS2 region, which is commonly used, as well as ITS1, in DNA metabarcoding studies of fungal diversity. To a large extent, ITS1 and ITS2 have similar results when used as DNA metabarcodes for fungi [15–17]. However, the ITS2 region lacks the insertions commonly found in ITS1 and thus reduces length variation [18]. This is important, as length variation can bias community pyrosequencing toward shorter amplicons. Also, ITS2 is the best-represented fungal genomic element in the public databases [19, 20]. Therefore, in studies similar to our project, use of ITS1 obtained with fungal specific primer (ITS1F) can be helpful in eliminating plant ITS amplification, and may turn out to be the method of choice in cases of mixed plant and fungal genomic DNA [21]. However, it is necessary to take into account that ITS1F (with constricted, specific range toward exclusion of all eukarya except fungal taxa), may not be able to amplify several fungal taxa because it is hampered with a high degree of mismatches relative to the target sequences [22, 23].
The ITS2 region was amplified with eukaryote-specific ITS3 and ITS4 primers (ITS3: GCATCGATGAAGAACGCAGC; ITS4: TCCTCCGCTTATTGATATGC) [22, 24]. Multiplex identifiers (MIDs) were attached to the primers’ ends to carry out in consequence the simultaneous analysis of all samples.
The amplicon library pool was sequenced with 454 pyrosequencing on the GS Junior sequencer (Roche, USA) according to the recommendations of the manufacturer [25]. The ITS2 locus reads were processed by QIIME Version 1.6.0 (Quantitative Insights into Microbial Ecology) [26]. To reduce the amount of erroneous sequences and thus increase the accuracy of the whole pipeline, the denoising procedure was employed [27].
Next steps included assigning multiplexed reads to samples based on their specific MIDs (demultiplexing), removing the low-quality or ambiguous reads, truncating primers, and other accessorial sequences. Chimeric sequences were detected using the UCHIME algorithm with the Unite database [28–30]. All of the reads were clustered into operational taxonomic units (OTUs) at 97% sequence similarity using the UCLUST method [31]. Representative sequences were chosen according to their abundance between similar reads. Low-abundance OTUs, which have less than four copies (singletons, doubletons and tripletons), were deleted over all of the analysis [32]. All 454 pyrosequencing data of the present investigation are available through the Sequence Read Archive (SRA) under BioProject PRJNA353503, with run accession numbers from SRR5022991 to SRR5023010 [33].
Phylogenetic and statistical analyses. Taxonomical identification of representative sequences was carried out by the BLAST method using Genbank databases [34, 35]. Query coverage ≥ 99% was recognized as significant. Query identity of ≥ 99% was considered identification at the species level; identity of ≥ 98–95% was considered reliable identification at the genus level. The smaller similarity Ribosomal Database Project classifier, along with the Unite database (minimum confidence at 0.9), were implemented to assign OTUs to a higher taxonomic rank [29, 30, 36, 37].
Alignment of representative sequences was carried out using MAFFT algorithm G-INS-1 [38]. A phylogenetic tree was conducted with MEGA5 using the Maximum Likelihood method, based on the Tamura-Nei model with 1000 bootstrap replicates [39–41].
Vegdist and hclust R functions were used for computing Bray-Curtis dissimilarity indices and UPGMA hierarchical clustering of OTUs, showing their coexistence in the samples [42]. Heatmaps were generated with QIIME 1.8.0 with log-transformed abundance data. OTUs were sorted by phylogenetic or hierarchical trees.
Beta diversity between samples was calculated by beta_diversity.py script in QIIME with unweighted UniFrac metric [26, 43]. To check the robustness of estimated beta diversity, jackknifed analysis, with 96 reads per sample depth and 100 replicates, was performed. The results were visualized with Principal coordinate analysis (PCoA) in a 2D scale plot.
Bray-Curtis, weighted and unweighted UniFrac dissimilarity indices were used for measuring the strength and significance of sample groupings with Permutational Multivariate Analysis of Variance (PERMANOVA) and Analysis of Similarity (ANOSIM) with script compare_categories.py [26].
Agar plate-based method of isolation and identification of fungi. Representative subsamples (200 grains) of each cultivar were surface-sterilized by being shaken for 2 min in 5% sodium hypochlorite. Then they were rinsed twice in sterilized water. The surfacesterilized grains were placed on 90 mm Petri dishes (10 grains per dish) with potato-sucrose agar (PSA) and incubated at 24°C for 10–14 days. The isolated fungal colonies from every grain were identified by visual and microscopic observations according to Ellis, Gerlach and Nirenberg, Lawrence, Rotondo, and Gannibal, and Samson et al. [44–47]. To present data comparable to those obtained with NGS, relative abundance was calculated as the number of all isolates of a certain taxon divided by the total number of fungal isolates (%). A more conventional index of seed expertise, infection frequency, was calculated as the number of grains infected by the fungus (%).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
NGS-based identification of fungi. After quality filtering and removal of nonfungal and chimeric sequences, in total, 8484 fungal reads were obtained and clustered into 43 operational taxonomic units (OTUs). The number of observed OTUs in the grain samples was ranged from 10 to 27. The estimated OTU richness was higher on the s urface ( Chao1 = 2 4.3 ± 4 .4, A CE = 2 6.2 ± 4 .9 (2014); C hao1 = 3 0.7 ± 4 .2, A CE = 2 9.7 ± 1 .8 (2015)), than inside (Chao1 = 15.1 ± 1.5, ACE = 16.8 ± 1.9 (2014); Chao1 = 19.3 ± 2.0, ACE = 21.7 ± 1.7 (2015)) the barley grains. The rarefaction curves pointed that the diversity of some samples might be underestimated, although all rarefaction curves were beyond the linear ranges.
All of the OTUs were assigned to Basidiomycota and Ascomycota phyla (Fig. 1).
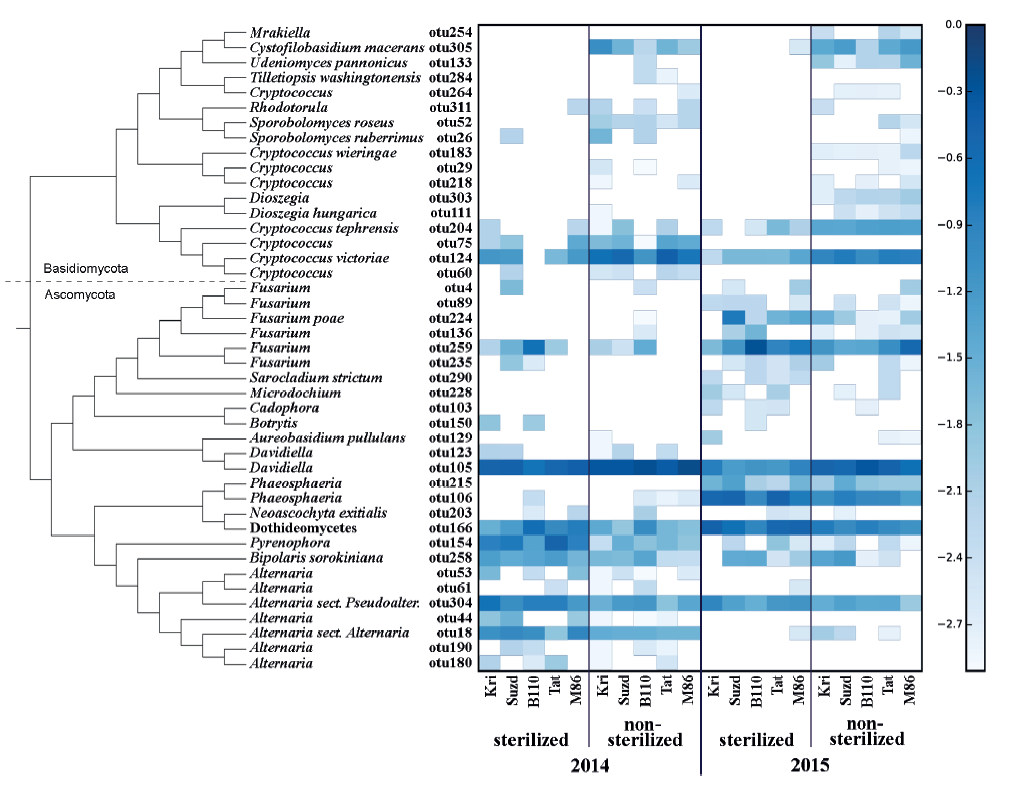
One of the most abundant OTU, otu166, assigned as Dothideomycetes, failed to be identified to a more precise taxonomical level. It has similar characteristics (BLASTn 100% query coverage and identity) with several GenBank sequences (e.g., EU552134, AJ279448, HG935454) which can be joined together only at the rank of class. More likely, it coincides with Epicoccum nigrum, the one abundant Dothideomycete identified during mycological analysis.
Eleven minor OTUs (Alternaria related otu44, otu53, otu61, otu180, and otu190; Cryptococcus related otu29, otu60, otu218, and otu264; Davidiella related otu123; and Fusarium related otu89) had no close relation to any known species but appeared in the same samples where a major OTU of a certain genus was abundant. However, potentially such satellite OTUs represent rare and/or poorly studied species, but most likely they are technical errors or sequence variances, which can occur despite all filtering and trimming procedures.
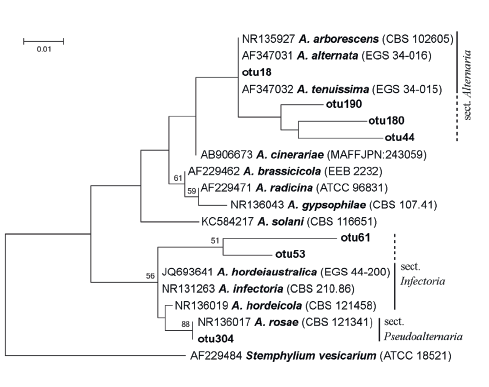
From seven clustered OTUs that were assigned as Alternaria, the most abundant OTUs, otu18 and otu304, can refer to Alternaria and Pseudoalternaria sections respectively (Fig. 3) [46, 48].
From ITS2 sequences combined into six OTUs and designated as Fusarium, several OTUs can be readily assigned as two synapomorphic (Fig. 4) clades, similar to that described by Watanabe et al. [49]. Two OTUs (F. poae – otu224 and Fusarium sp. – otu259) were abundant, but four OTUs appeared as solitary sequences.
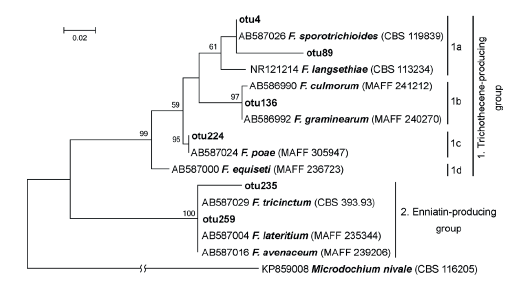
Distribution of Fusarium-related OTUs among clusters corresponded to the prevalent toxic secondary metabolite production. The first cluster consisted of Fusarium fungi that are able to produce the trichothecene group metabolites. The subcluster 1a included F. sporotrichioides and F. langsethiae, which are the main producers of type A trichothecenes (like T-2 and HT-2 toxins); the subcluster 1b included F. culmorum and F. graminearum the producers of type B trichothecenes (like DON, or NIV). Species F. poae (subcluster 1c) produces trichothecenes of types A and B, and enniatins (ENNs). Fungus F. equiseti (subcluster 1d) is able to produce ENNs, but according to some authors, it can also produce a small amount of type A trichothecenes [50, 51]. The subcluster 2 brought together Fusarium fungi that are able to synthesize ENNs: F. tricinctum, F. avenaceum, and F. lateritium [52].
Fourteen OTUs that were defined to the species level belonged to Bipolaris sorokiniana, Fusarium poae, Neoascochyta exitialis, Sarocladium strictum, Cystofilobasidium macerans, Udeniomyces pannonicus, Cryptococcus victoriae, Cryptococcus tephrensis, Cryptococcus wieringae, Sporobolomyces ruberrimus, Sporobolomyces roseus, Dioszegia hungarica, Aureobasidium pullulans, and Tilletiopsis washingtonensis.
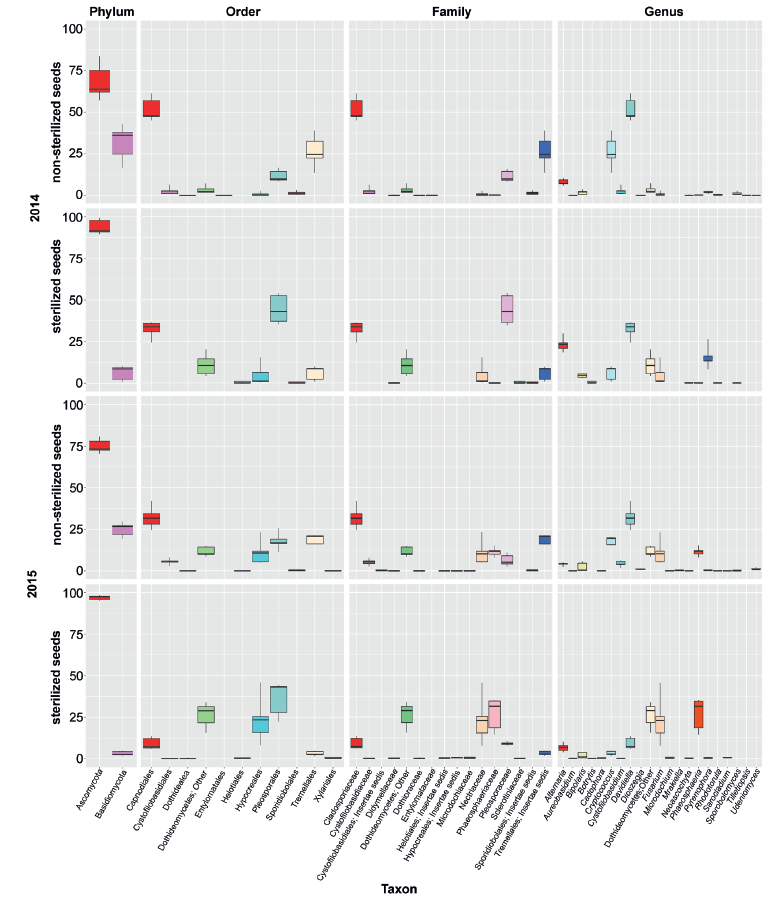
In general, the mycobiome of nonsterilized barley grains was characterized by a greater abundance of Basidiomycetes in comparison with surface-sterilized grains. The most abundant fungi in nonsterilized grains were Davidiella (Cladosporium) spp. and Cryptoccocus spp. After surface sterilization, the average abundance of Fusarium, Alternaria, Pyrenophora, and Phaeosphaeria, as well as fungi from Dothideomycetes, increased, but the ratio of those taxa depended on the year.
Comparison of taxonomical structure and relative abundance between groups of samples combined by crop year (2014/2015) and type of treatment (sterilized/non-sterilized) reflected significant distinctions in both cases (Fig. 5). Nevertheless, distinctions between sterilized and non-sterilized grain mycobiota (ANOSIM R = 0.64, 0.76, 0.69; P = 0.001, 0.001, 0.001; PERMANOVA pseudo F = 9.89, 17.7, 10.93; P = 0.001, 0.001, 0.001; data shown successively for Bray-Curtis, Weighted and Unweighted UniFrac community dissimilarity matrices) occurred to be more strong, than those determined in successive crop years (ANOSIM R = 0.53, 0.21, 0.37; P = 0.001, 0.02, 0.006; PERMANOVA pseudo F = 8.03, 4.97, 5.02, P = 0.001, 0.019, 0.003).
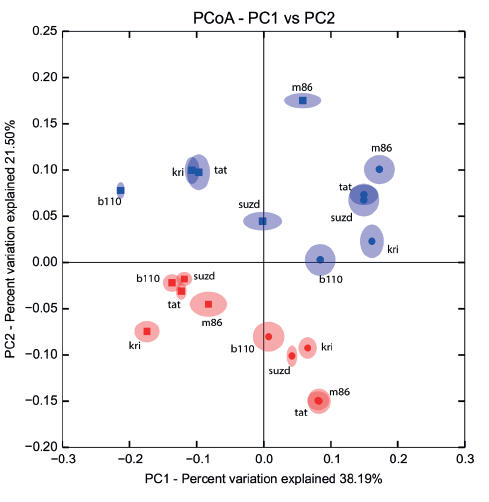
The fungal species composition of non-sterilized grains differed from the mycobiome of surfacesterilized grains primary due to a higher abundance of Basidiomycetes (Cryptococcus spp. and other Tremellales, and Cystofilobasidium macerans and other Cystofilobasidiaceae) and Davidiella (Cladosporium spp.) in the non-sterilized grains. All Basidiomycetes disappeared or became sparse after surface sterilization. The most abundant of them, Cryptococcus tephrensis (otu204) and C. victoriae (otu124), were also revealed inside grains but in fewer samples and in lesser amounts. Several OTUs, e.g., Cryptococcus wieringae (otu183), Mrakiella sp. (otu254), and Dioszegia sp. (otu303), tended to present on seed surfaces during only one year. Mycobiomes observed in two different growing seasons differed by abundance of Pyrenophora sp. in 2014 and Fusarium spp. and Phaeosphaeria sp. in 2015. The Alternaria, Bipolaris, and Epicoccum genera were relatively abundant in both sample sets. More detailed results of fungal coexistence in the samples are introduced in Fig. 6.

Agar plate-based method of identification of fungi. From 87 to 117 fungal isolates per 100 grains were obtained from each sample. As a result of two growing seasons, a total of 18 taxa of seed-borne fungi were identified (Table 1). The taxonomic position of some fungi was vague due to the lack of sporulation (Mycelia sterilia). In both years, the appearance of Alternaria, Bipolaris, Epicoccum, and Fusarium species was common. Species of the genus Pyrenophora were found only in the 2014 growing season. Some potentially toxigenic fungi, such as Penicillium, Aspergillus, Cladosporium and unidentified Zygomycota, were found only in a few samples. No Basidiomycetes were isolated and identified by agar plate-based assay.
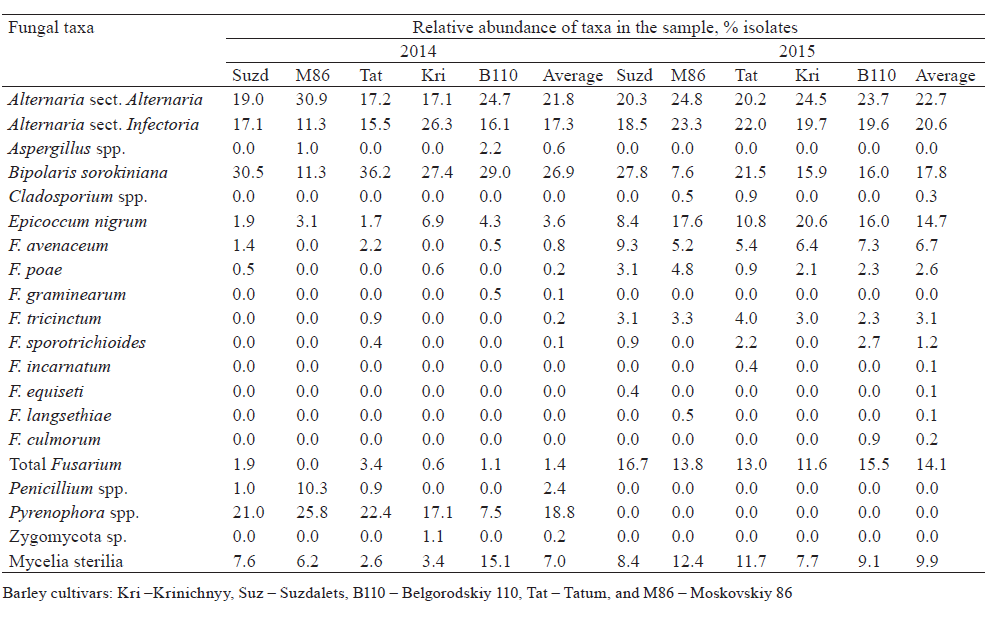
In both years, fungi of the genus Alternaria predominated in barley grain samples. The members of two sections, Alternaria and Infectoriae, were determined. More precise identification was not performed, since species concept is debatable for Alternaria and Infectoriae [53–56] sections.
Contamination of the barley grains by Fusarium spp. varied significantly in 2014 and 2015. In 2014, the Fusarium infection frequency was low (0–4%) and represented by five species, of which F. avenaceum was the most frequent (infection frequency up to 2.5%, relative abundance of isolates up to 2.2%). In 2015, the infection of barley grains with Fusarium spp. was considerably higher (infection frequency 14–19%, isolate abundance 12–17%). Eight Fusarium species were identified; four of them were common for both years.
Comparison of methods. In total, 43 OTUs assigned as Ascomycota (26) and Basidiomycota (17) were revealed by DNA metabarcoding. Only 14 OTUs were assigned to species level. From those species, only two were reoccurred in traditional mycological analysis. The other 12 species either were not detected among isolates grown up from grains on agar medium or were Mycelia sterilia. At the same time, the conventional mycological seed test revealed 17 Ascomycetes, including 11 species, apart from some Zygomycetes and sterile Ascomycetes. Basidiomycetes were not recovered by conventional assay. Two species (Fusarium poae and Bipolaris sorokiniana), one section (Alternaria sect. Alternaria), and three genera (Davidiella [Cladosporium], Fusarium, and Pyrenophora) were formally common for both assays. In general, the list of undoubtedly identified dominant taxa coincides with the results of the NGS mycobiome study of Canadian barley grains [3].
The predominant OTUs from Alternaria were identified as Alternaria and Pseudoalternaria sections when Alternaria and Infectoriae sections were fixed during mycological analysis. In both cases, taxa were identified to the section level. Such precision is sufficient for the majority of practical purposes, e.g., for tests of seed, food, or feed-grain quality. The big section Infectoriae and lately described section Pseudoalternaria are morphologically similar and phylogenetically close groups [46]. This obviously can be the cause of errors, if identification is based on morphological features.
Both methods similarly reflected a very low abundance of Fusarium spp. in 2014 and a higher quantity in 2015. Traditional mycological analysis revealed nine Fusarium species. DNA metabarcoding results were more limited; only one OTU was identified as a certain species, F. poae, but the others were assigned to a clade level. Phylogenetic resolution derived from ITS2 is not useful in defining Fusarium species. Recently, Fusarium-specific primers targeting translation elongation factor 1 (TEF1) were evaluated and successfully applied to analyze Fusarium communities in soil and plant material [57].
The taxonomy of Fusarium fungi is confusing and various classification systems have been proposed [58]. For Fusarium, chemotaxonomy is considered a supplement to traditional morphology-based taxonomy. Several fungal genes involved in trichothecene and enniatins biosynthesis have been defined and used for development of molecular assays aimed at identification. In spite of the ITS sequences used in our analysis, the results strongly suggested the division of fungi based on their ability to produce metabolites. In the future, this will provide an opportunity to predict the severity of grain contamination by some mycotoxins according to the number of certain identified OTUs.
Fusarium avenaceum, F. poae, F. tricinctum, and F. sporotrichioides were the most a bundant representatives of the genus. They are the typical pathogens of barley in northwestern Russia [59, 60]. Most likely, multi-copy otu259 discovered by DNA metabarcoding is associated with F. avenaceum, which occurred frequently on the barley grain.
Both methods revealed pathogenic fungi from Pleosporaceae: Bipolaris and Pyrenophora. Those fungi have different patterns of appearance through the cropping seasons. DNA metabarcoding demonstrated higher sensitivity. Pyrenophora sp. colonies were not recovered in 2015 at all, but several respective reads were obtained for 7 out of 10 samples.
Davidiella (Cladosporium) associated reads were abundant in DNA metabarcoding assay in non-sterilized samples but only single colonies were detected in the agar plate-based test. Underestimation of relative abundance of the fungus in the latter case can be result of two reasons: rapidly spreading colonies suppress or mask slowly growing fungi, and infected individual grains contain not uniform quantity of fungal biomass that appear as insufficient correlation between the number of infected grains and the amount of fungal DNA in the whole sample.
Four fungal genera revealed by only DNA metabarcoding contained agents of cereal diseases (Neoascochyta, Botrytis, Microdochium, and Phaeosphaeria). The first three taxa were represented by solitary reads. Phaeosphaeria (otu106 and otu215) were found in 14 of 20 samples. In 2015, in surface-sterilized samples, the relative abundance of Phaeosphaeria reads varied between 11 and 36%. Sequences of otu106 had the closest similarity (99%) with representative sequences of Parastagonospora avenae (Septoria avenae or Stagonospora avenae), widespread fungus causing leaf blotch of barley and some other cereals, and Parastagonospora poagena, a recently described fungus from Poa sp. [61, 62]. Less abundant OTU, otu215, had a similarity of 98%, with several Phaeosphaeria species and with some unidentified endophytes.
ВЫВОДЫ
DNA metabarcoding, based on high-throughput sequencing, is a sensitive and powerful method of grain mycobiome analysis that provides large amounts of data. However, at this time, not all fungi can be identified to species level by molecular markers, especially by rDNA sequences. In spite of universality, rDNA has a limitation as a taxonomic marker. The resolution of the ITS sequence-based method is not enough to differentiate many fungal species. For instance, many Fusarium species have nonorthologous copies of ITS2. Many other important plant pathogenic and toxigenic fungi also can be identified up to genus level, but that is not always informative in the framework of mycological seed expertise. Erroneous and chimerical sequences, as well as the lack of reference sequences of many species, still limits wide application of NGS-based technologies in biodiversity studies.
The most complete and credible results can be obtained when several approaches are implemented simultaneously. Combining the results of DNA metabarcoding and traditional culture-plating assay allowed us to revise the diversity of fungi colonizing on the surface of and inside barley grains in Leningradskaya oblast (northwest Russia).
Fungal species diversity of barley grain revealed by DNA metabarcoding formally exceeded the traditional microbiological culture-based agar plating: 43 operational taxonomic units (OTUs) vs. 17 taxa of genus or species level. DNA metabarcoding assay allowed seven ascomycete taxa to be added to the total list. Of those additional taxa, only Phaeosphaeria was abundant internal fungus. Seventeen OTUs belonging mainly to surface-seed-borne, yeastlike Basidiomycetes were completely outside the scope of traditional analysis. Meanwhile, routine mycological analysis, in contrast to DNA metabarcoding, resulted in precise identification of practically important Fusarium species. On the other side, due to DNA analysis, one Alternaria taxon was reidentified as Alternaria section Pseudoalternaria instead of section Infectoriae.
КОНФЛИКТ ИНТЕРЕСОВ
The authors declare that there is no conflict of interest regarding the publication of this article.БЛАГОДАРНОСТИ
The authors are grateful to A. Vagin (Volosovo State Seed-Trial Ground, Russia) for providing seed samples and key information on them, and to M. Gomzhina (All-Russian Institute of Plant Protection) for technical assistance. Pyrosequencing was conducted using the equipment of the Core Centrum “Genomic Technologies, Proteomics and Cell Biology” at All-Russia Research Institute for Agricultural Microbiology (ARRIAM, St. Petersburg, Russia).ФИНАНСИРОВАНИЕ
This work was supported financially by the Russian Science Foundation (RSF) (№ 19-76-30005).СПИСОК ЛИТЕРАТУРЫ
- Food and Agriculture Organization of the United Nations [Internet]. [cited 2018 Oct 15]. Available from: http://faostat3.fao.org.
- Arendt E, Zannini E. Cereal grains for the food and beverage industries. Cambridge: Woodhead Publishing; 2013. 512 p. DOI: https://doi.org/10.1533/9780857098924.
- Chen W, Turkington TK, Levesque CA, Bamforth JM, Patrick SK, Lewis CT, et al. Geography and agronomical practices drive diversification of the epiphytic mycoflora associated with barley and its malt end product in western Canada. Agriculture Ecosystems and Environment. 2016;226:43–55. DOI: https://doi.org/10.1016/j.agee.2016.03.030.
- Flannigan B. The microbiota of barley and malt. In: Priest FG, Campbell I, editors. Brewing microbiology. Boston: Springer; 2003. pp. 113–180. DOI: https://doi.org/10.1007/978-1-4419-9250-5_4.
- Buee M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, et al. 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytologist. 2009;184(2):449–456. DOI: https://doi.org/10.1111/j.1469-8137.2009.03003.x.
- Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R, et al. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Applied and Environmental Microbiology. 2007;73(21):7059–7066. DOI: https://doi.org/10.1128/AEM.00358-07.
- Jumpponen A. Soil fungal communities underneath willow canopies on a primary successional glacier forefront: rDNA sequence results can be affected by primer selection and chimeric data. Microbial Ecology. 2007;53(2):233–246. DOI: https://doi.org/10.1007/s00248-004-0006-x.
- Kuramae EE, Verbruggen E, Hillekens R, de Hollander M, Roling WFM, van der Heijden MGA, et al. Tracking fungal community responses to maize plants by DNA- and RNA-based pyrosequencing. PLoS ONE. 2013;8(7). DOI: https://doi.org/10.1371/journal.pone.0069973.
- Links MG, Demeke T, Grafenhan T, Hill JE, Hemmingsen SM, Dumonceaux TJ. Simultaneous profiling of seedassociated bacteria and fungi reveals antagonistic interactions between microorganisms within a shared epiphytic microbiome on Triticum and Brassica seeds. New Phytologist. 2014;202(2):542–553. DOI: https://doi.org/10.1111/nph.12693.
- Yildirim EA, Laptev GYu, Il’ina LA, Nikonov IN, Filippovav VA, Soldatova VV, et al. The investigation of endophytic microorganisms as a source for silage microbiocenosis formation using NGS-sequencing. Agricultural Biology. 2015;50(6):832–838. DOI: https://doi.org/10.15389/agrobiology.2015.6.832eng.
- Igiehon NO, Babalola OO. Biofertilizers and sustainable agriculture: exploring arbuscular mycorrhizal fungi. Applied Microbiology and Biotechnology. 2017;101(12):4871–4881. DOI: https://doi.org/10.1007/s00253-017-8344-z.
- Galazka A, Grzadziel J. Fungal genetics and functional diversity of microbial communities in the soil under long-term monoculture of maize using different cultivation techniques. Frontiers in Microbiology. 2018;9. DOI: https://doi.org/10.3389/fmicb.2018.00076.
- Mancini V, Murolo S, Romanazzi G. Diagnostic methods for detecting fungal pathogens on vegetable seeds. Plant Pathology. 2016;65(5):691–703. DOI: https://doi.org/10.1111/ppa.12515.
- Nicolaisen M, Justesen AF, Knorr K, Wang J, Pinnschmidt HO. Fungal communities in wheat grain show significant co-existence patterns among species. Fungal Ecology. 2014;11:145–153. DOI: https://doi.org/10.1016/j.funeco.2014.06.002.
- Bazzicalupo AL, Balint M, Schmitt I. Comparison of ITS1 and ITS2 rDNA in 454 sequencing of hyperdiverse fungal communities. Fungal Ecology. 2013;6(1):102–109. DOI: https://doi.org/10.1016/j.funeco.2012.09.003.
- Blaalid R, Kumar S, Nilsson RH, Abarenkov K, Kirk PM, Kauserud H. ITS1 versus ITS2 as DNA metabarcodes for fungi. Molecular Ecology Resources. 2013;13(2):218–224. DOI: https://doi.org/10.1111/1755-0998.12065.
- Mello A, Napoli C, Murat C, Morin E, Marceddu G, Bonfante P. ITS-1 versus ITS-2 pyrosequencing: a comparison of fungal populations in truffle grounds. Mycologia. 2011;103(6):1184–1193. DOI: https://doi.org/10.3852/11-027.
- Martin KJ, Rygiewicz PT. Fungal-specific PCR primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiology. 2005;5. DOI: https://doi.org/10.1186/1471-2180-5-28.
- Kostovcik M, Bateman CC, Kolarik M, Stelinski LL, Jordal BH, Hulcr J. The ambrosia symbiosis is specific in some species and promiscuous in others: evidence from community pyrosequencing. ISME Journal. 2015;9(1):126–138. DOI: https://doi.org/10.1038/ismej.2014.115.
- Nilsson RH, Ryberg M, Abarenkov K, Sjokvist E, Kristiansson E. The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiology Letters. 2009;296(1):97–101. DOI: https://doi.org/10.1111/j.1574-6968.2009.01618.x.
- Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes – application to the identification of mycorrhizae and rusts. Molecular Ecology. 1993;2(2):113–118. DOI: https://doi.org/10.1111/j.1365-294X.1993.tb00005.x.
- Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiology. 2010;10. DOI: https://doi.org/10.1186/1471-2180-10-189.
- De Beeck MO, Lievens B, Busschaert P, Declerck S, Vangronsveld J, Colpaert JV. Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS ONE. 2014;9(6). DOI: https://doi.org/10.1371/journal.pone.0097629.
- White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: a guide to methods and applications. Orlando: Academic Press; 1990. pp. 315–322. DOI: https://doi.org/10.1016/B978-0-12-372180-8.50042-1.
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. DOI: https://doi.org/10.1038/nature03959.
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7(5):335–336. DOI: https://doi.org/10.1038/nmeth.f.303.
- Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nature Methods. 2010;7(9):668–669. DOI: https://doi.org/10.1038/nmeth0910-668b.
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. DOI: https://doi.org/10.1093/bioinformatics/btr381.
- Abarenkov K, Nilsson RH, Larsson KH, Alexander IJ, Eberhardt U, Erland S, et al. The UNITE database for molecular identification of fungi – recent updates and future perspectives. New Phytologist. 2010;186(2):281–285. DOI: https://doi.org/10.1111/j.1469-8137.2009.03160.x.
- Koljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, et al. UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytologist. 2005;166(3):1063–1068. DOI: https://doi.org/10.1111/j.1469-8137.2005.01376.x.
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. DOI: https://doi.org/10.1093/bioinformatics/btq461.
- Lindahl BD, Nilsson RH, Tedersoo L, Abarenkov K, Carlsen T, Kjoller R, et al. Fungal community analysis by highthroughput sequencing of amplified markers – a user’s guide. New Phytologist. 2013;199(1):288–299. DOI: https://doi.org/10.1111/nph.12243.
- Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Research. 2011;39:D19–D21. DOI: https://doi.org/10.1093/nar/gkq1019.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215(3):403–410. DOI: https://doi.org/10.1016/S0022-2836(05)80360-2.
- Genbank [Internet]. [cited 2018 Oct 15]. Available from: https://www.ncbi.nlm.nih.gov/genbank.
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology. 2007;73(16):5261–5267. DOI: https://doi.org/10.1128/AEM.00062-07.
- Unite community [Internet]. [cited 2018 Oct 15]. Available from: https://unite.ut.ee.
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research. 2002;30(14):3059–3066. DOI: https://doi.org/10.1093/nar/gkf436.
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011;28(10):2731–2739. DOI: https://doi.org/10.1093/molbev/msr121.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution. 2013;30(12):2725–2729. DOI: https://doi.org/10.1093/molbev/mst197.
- Tamura K, Nei M. estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Molecular Biology and Evolution. 1993;10(3):512–526.
- The R project for statistical computing [Internet]. [cited 2018 Oct 15]. Available from: http://www.R-project.org/.
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology. 2005;71(12):8228–8235. DOI: https://doi.org/10.1128/AEM.71.12.8228-8235.2005.
- Ellis MB. Dematiaceous hyphomycetes. Surrey: CMI, Kew; 1971. 608 p.
- Gerlach W, Nirenberg H. The genus Fusarium – a pictorial atlas. Mitteilungen der Biologischen Bundesanstalt für Land – und Forstwirtschaft. 1982;209:1–406.
- Lawrence DP, Rotondo F, Gannibal PB. Biodiversity and taxonomy of the pleomorphic genus Alternaria. Mycological Progress. 2016;15(1). DOI: https://doi.org/10.1007/s11557-015-1144-x.
- Samson RA, Hoekstra ES, Frisvad JC, Filtenborg O. Introduction to Food- and Airborne Fungi. Utrecht: Centraalbureau voor Schimmelcultures; 2002. 389 p.
- Lawrence DP, Gannibal PB, Peever TL, Pryor BM. The sections of Alternaria: formalizing species-group concepts. Mycologia. 2013;105(3):530–546. DOI: https://doi.org/10.3852/12-249.
- Watanabe M, Yonezawa T, Lee K, Kumagai S, Sugita-Konishi Y, Goto K, et al. Molecular phylogeny of the higher and lower taxonomy of the Fusarium genus and differences in the evolutionary histories of multiple genes. BMC Evolutionary Biology. 2011;11. DOI: https://doi.org/10.1186/1471-2148-11-322.
- Thrane U. Developments in the taxonomy of Fusarium species based on secondary metabolites. In: Summerell BA, editor. Fusarium: Paul E. Nelson memorial symposium. St. Paul: APS Press; 2001. pp. 29–49.
- Yli-Mattila T, Gagkaeva TYu. Fusarium toxins in cereals in Northern Europe and Asia. In: Deshmukh SK, Misra JK, Tewari JP, Papp T, editors. Fungi: applications and management strategies. Boca Raton: CRC Press; 2016. pp. 293–317.
- Jestoi M. Emerging Fusarium-mycotoxins fusaproliferin, beauvericin, enniatins, and moniliformin – A review. Critical Reviews in Food Science and Nutrition. 2008;48(1):21–49. DOI: https://doi.org/10.1080/10408390601062021.
- Gannibal PB. Distribution of Alternaria species among sections. 2. Section Alternaria. Mycotaxon. 2015;130(4):941–949. DOI: https://doi.org/10.5248/130.941.
- Woudenberg JHC, Seidl MF, Groenewald JZ, de Vries M, Stielow JB, Thomma B, et al. Alternaria section Alternaria: Species, formae speciales or pathotypes? Studies in Mycology. 2015(82):1–21. DOI: https://doi.org/10.1016/j.simyco.2015.07.001.
- Andersen B, Sorensen JL, Nielsen KF, van den Ende BG, de Hoog S. A polyphasic approach to the taxonomy of the Alternaria infectoria species-group. Fungal Genetics and Biology. 2009;46(9):642–656. DOI: https://doi.org/10.1016/j.fgb.2009.05.005.
- Gannibal PB, Lawrence DP. Distribution of Alternaria species among sections. 3. Sections Infectoriae and Pseudoalternaria. Mycotaxon. 2016;131(4):781–790. DOI: https://doi.org/10.5248/131.781.
- Karlsson I, Edel-Hermann V, Gautheron N, Durling MB, Kolseth AK, Steinberg C, et al. Genus-specific primers for study of Fusarium communities in field samples. Applied and Environmental Microbiology. 2016;82(2):491–501. DOI: https://doi.org/10.1128/AEM.02748-15.
- Leslie JF, Summerell BA. The Fusarium laboratory manual. Oxford: Blackwell Publishing; 2006. 388 p.
- Gagkaeva TYu, Gavrilova OP, Levitin MM, Novozhilov KV. Fuzarioz zernovykh kulʹtur [The fusariosis of cereal crops]. Zashchita i karantin rasteniy [Plant protection and quarantine]. 2011;(5):69–120. (In Russ.).
- Gavrilova OP, Gagkaeva TYu, Burkin AA, Kononenko GP. Mycological infection by Fusarium strains and mycotoxins contamination of oats and barley in the north of nonchernozem’e. Agricultural biology. 2009;44(6):89–93. (In Russ.).
- Quaedvlieg W, Verkley GJM, Shin HD, Barreto RW, Alfenas AC, Swart WJ, et al. Sizing up Septoria. Studies in Mycology. 2013(75):307–390. DOI: https://doi.org/10.3114/sim0017.
- Crous PW, Shivas RG, Quaedvlieg W, van der Bank M, Zhang Y, Summerell BA, et al. Fungal Planet description sheets: 214–280. Persoonia. 2014;32:184–306. DOI: https://doi.org/10.3767/003158514X682395.
